Die Ergebnisse zeigen Tendenz zu geringeren Toxizitäten und besseren Ergebnissen bei Patienten, die eine Kombination aus ONIVYDE/5-FU und gezielter Anti-IL-1alpha-Therapie erhalten
AUSTIN, Texas, June 19, 2024 (GLOBE NEWSWIRE) -- XBiotech (NASDAQ: XBIT) hat heute Daten aus seiner randomisierten, doppelt verblindeten, placebokontrollierten multizentrischen Phase-I/II-Studie zur Behandlung von fortgeschrittenem Bauchspeicheldrüsenkrebs bekanntgegeben. Die als 1-BETTER bekannte Studie untersuchte den Antikörper Natrunix (Anti-Interleukin-1alpha) in Kombination mit einem etablierten Chemotherapieschema (ONIVYDE (ON) + 5-Fluorouracil (5FU) + Leucovorin (LV)), einem Schema, das bereits in großem Umfang zur Behandlung von Bauchspeicheldrüsenkrebs eingesetzt wird, aber mit schwierigen Toxizitäten und weniger als idealen Überlebensergebnissen verbunden ist. Natrunix wurde als Anti-Krebs-Wirkstoff für den Einsatz in zytotoxischen Chemotherapie-Kombinationen untersucht, wo es nach Ansicht des Unternehmens möglicherweise auch die Verträglichkeit der Chemotherapie verbessern könnte.
Der Phase-I-Teil war eine Dosiseskalationsstudie bei Patienten mit metastasiertem Bauchspeicheldrüsenkrebs, um festzustellen, ob dosislimitierende Toxizitäten (DLT) in Kombination mit dem ON+5FU+LV-Schema in der Zweit- oder Drittlinienbehandlung auftreten. DLT wurden bei Natrunix nicht erwartet, und es wurden auch keine beobachtet. Die Natrunix-Dosis im Phase-II-Teil war also die höchste Dosis, die im Phase-I-Teil verwendet wurde.
65 Teilnehmer wurden im Verhältnis 1:1 in die Phase-II-Studie randomisiert und erhielten entweder Natrunix+ ON+5FU+LV (Arm1) oder Placebo+ON+5FU+LV (Arm2). Es wurden 33 Teilnehmer in Arm1 und 32 in Arm2 aufgenommen. Die Phase-II-Behandlung dauerte 24 Wochen, wobei die Teilnehmer über einen Zeitraum von insgesamt 12 Zyklen jede zweite Woche eine Therapie erhielten.
Die in die Studie eingeschlossenen Teilnehmer hatten ein bestätigtes metastasiertes, inoperables oder rezidivierendes Adenokarzinom des exokrinen Pankreas und mussten nach einer vorherigen Gemcitabin-basierten Therapie oder einer FOLFIRINOX- und Gemcitabin-haltigen Therapie ein Fortschreiten der Erkrankung erlitten haben. Alle Patienten mussten mindestens eine messbare Läsion gemäß den Response Evaluation Criteria in Solid Tumors (RECIST v1.1) aufweisen.
Der primäre Endpunkt der Phase-II-Studie war die Bewertung der Sicherheit und Verträglichkeit von Natrunix in Verbindung mit der Kombination ON+5FU+LV. Insgesamt traten während der 24-wöchigen Behandlungsdauer in der Natrunix-Gruppe im Vergleich zu Placebo weniger unerwünschte Ereignisse (UE) (297 ggü. 336) auf, wobei die Zahl der Ereignisse in bestimmten Kategorien von unerwünschten Ereignissen während dieser Zeit deutlich geringer war. Die Zahl der Teilnehmer, bei denen während der 24-wöchigen Behandlung schwerwiegende unerwünschte Ereignisse (SUE) auftraten, war in der Natrunix-Gruppe (9 von 33) um 28 % geringer als bei Placebo (12 von 32). Bei Teilnehmern, die Natrunix+ON+5FU+LV-Kombination erhielten, verringerte sich die Zahl der Krankenhausaufenthalte (80 Tage gegenüber 120 Tagen) während des 24-wöchigen Behandlungszeitraums um etwa 33 % im Vergleich zu Teilnehmern, die Placebo+ON+5FU+LV-Kombination erhielten.
Teilnehmer, die Natrunix-Kombination erhielten, berichteten am letzten Tag des 24-wöchigen Behandlungszeitraums im Vergleich zu Teilnehmern, die Placebo+ON+5FU+LV-Kombination erhielten, auch über eine um 22 % geringere Müdigkeit (28 ggü. 36), einen um 32 % verbesserten Appetit (19 ggü. 28) und um 41 % geringere Schmerzen (17 ggü. 29).
Schwere Durchfälle, die lebensbedrohlich sein können, sind eine erhebliche Komplikation bei der ON+5FU+LV-Therapie. Bei Patienten, die Kombination Natrunix+ON+5FU+LV im Vergleich zu Placebo+ON+5FU+LV erhielten, war die Häufigkeit schwerer Durchfälle während des 24-wöchigen Behandlungsschemas um das Zweifache reduziert (9 % gegenüber 19 %).
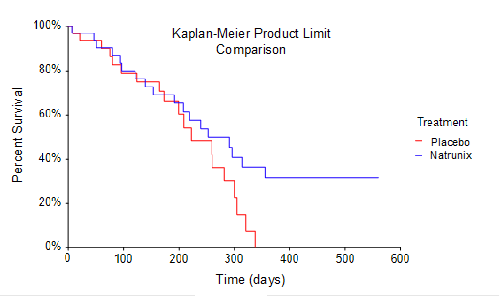
Das Gesamtüberleben (Overall Survival, OS), einer der sekundären Endpunkte der Phase-II-Studie, wurde konventionell als Zeit von der Randomisierung bis zum Tod definiert. Die Stichprobengröße für die Studie umfasste eine Intent-to-Treat-Analyse von 33 Teilnehmern, die in den Natrunix+ON+5FU+LV-Arm randomisiert wurden, gegenüber 32 Teilnehmern im Placebo+ON+5FU+LV-Arm. Es wurde eine Kaplan-Meier-Überlebenskurve unter Verwendung einer Produktgrenzenvergleichsmethode erstellt. Diese Daten unterstreichen die Beobachtung, dass kein Teilnehmer in der Placebo+ON+5FU+LV-Gruppe (n=32) länger als 330 Tage überlebte, während 8 Teilnehmer in der Natrunix+ON+5FU+LV-Gruppe (n=33) an Tag 330 noch am Leben waren. In Anbetracht des geringen Stichprobenumfangs deutet der grenzwertig statistisch signifikante p-Wert von p = 0,096 auf eine verlängerte Überlebenszeit der Teilnehmer hin, die das Natrunix-Behandlungsschema erhielten.
Der leitende Prüfarzt der Studie, Dr. David J. Park, medizinischer Onkologe und medizinischer Direktor des Providence St. Jude Crosson Institute in Fullerton, Kalifornien, erklärte: "Die Behandlung von fortgeschrittenem Bauchspeicheldrüsenkrebs in der Zweit- und Drittlinienbehandlung stellt eine große Herausforderung dar, sowohl was die Toxizität als auch die Wirksamkeit betrifft. Die Beobachtung dieser Trends zu geringerer Toxizität und potenziellem Überlebensvorteil ist bemerkenswert, insbesondere angesichts der begrenzten Stichprobengröße. Die potenzielle Wechselwirkung zwischen verringerter Toxizität, längerer Behandlungsdauer und verbesserter Überlebensrate ist für Ärzte, die diese Patienten behandeln, intuitiv verständlich. Diese Erkenntnisse sind äußerst wichtig."
Obwohl nur eine relativ kleine Anzahl von Patienten mit Bauchspeicheldrüsenkrebs in den Phase-II-Teil der Studie aufgenommen wurde, zeigen die Ergebnisse nach Ansicht des Unternehmens bessere Ergebnisse für die Natrunix+ON+5FU+LV-Gruppe im Vergleich zur Kontrollgruppe. Das Unternehmen ist der Ansicht, dass die geringere Anzahl von schwerwiegenden und unerwünschten Ereignissen, die signifikante Verringerung der Krankenhausaufenthalte und die Verbesserung des Überlebens während der oben beschriebenen Zeiträume für jeden dieser Parameter darauf hindeuten, dass Natrunix einen bahnbrechenden Fortschritt für die Behandlung von Bauchspeicheldrüsenkrebs darstellen könnte.
Über XBiotech
XBiotech leistet Pionierarbeit bei der Entdeckung und Entwicklung von zielgerichteten Antikörpern auf der Grundlage seiner True Human-Technologie. Das Unternehmen hat es sich zur Aufgabe gemacht, die Art und Weise, wie Antikörpermedikamente entdeckt und vermarktet werden, neu zu überdenken, indem es seine robuste Pipeline wirklich natürlicher menschlicher Antikörper für die Behandlung schwerer Krankheiten wie Entzündungen.
Vorsichtshinweis zu zukunftsgerichteten Aussagen und Studienergebnissen
Diese Pressemitteilung enthält zukunftsgerichtete Aussagen, einschließlich Erklärungen zu den Überzeugungen und Erwartungen des Managements, die mit erheblichen Risiken und Unsicherheiten verbunden sind. Zukunftsgerichtete Aussagen unterliegen inhärenten Risiken und Ungewissheiten bei der Vorhersage zukünftiger Ergebnisse und Bedingungen, die dazu führen könnten, dass die tatsächlichen Ergebnisse erheblich von den in diesen zukunftsgerichteten Aussagen prognostizierten abweichen. Diese Risiken und Unwägbarkeiten unterliegen den Angaben, die im Abschnitt "Risikofaktoren" in einigen unserer SEC-Berichte aufgeführt sind. Zukunftsgerichtete Aussagen in dieser Pressemitteilung spiegeln den Stand zum Zeitpunkt der Veröffentlichung dieser Pressemitteilung wider. Wir übernehmen keine Verpflichtung, unsere zukunftsgerichteten Aussagen zu aktualisieren, sei es aufgrund neuer Informationen, zukünftiger Ereignisse oder aus anderen Gründen, die nach dem Datum dieser Pressemitteilung eintreten. Das Unternehmen gibt keine Zusicherungen in Bezug auf das Gesamtüberleben oder andere Metriken, die über die hier ausdrücklich erwähnten Zeiträume hinausgehen. Es kann nicht garantiert werden, dass die in dieser Pressemitteilung erwähnten Studienergebnisse in zukünftigen Studien reproduziert werden können oder dass Natrunix von der US-amerikanischen Food and Drug Administration oder einer anderen Behörde zugelassen werden wird.
Kontakt
Wenyi Wei
wwei@xbiotech.com
Tel. 737-207-4600
Ein Foto zu dieser Mitteilung ist verfügbar unter https://www.globenewswire.com/NewsRoom/AttachmentNg/7a17b3d3-b304-47ae-b1e7-0950f0301f12

